Research
Quantum Information Science
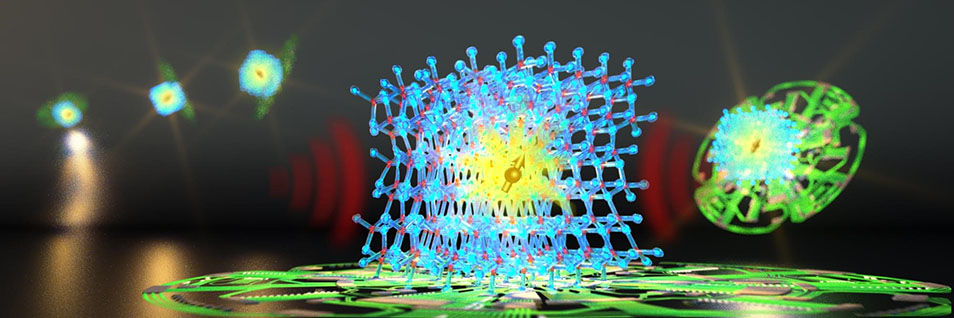
Study spin defects in wide-band gap semiconductors in search of promising systems for the realization of qubits in solid-state environments
The development of quantum technologies that can store and manipulate information has the potential to provide new paths for computing, communication, and sensing. Central to quantum information research is the development of quantum states that can be manipulated to perform tasks which cannot be emulated by ordinary counterparts, thanks to quantum processes that have no classical analog. Ground-breaking discoveries with the potential to transform computing technologies or generate a new class of nanoscale sensors with enhanced sensitivity require a detailed understanding and control of materials with robust quantum properties.
Units of quantum information - qubits - encoded by electronic states of defects in semiconductors are promising candidates because they combine the quantum properties of isolated atoms with the convenience and scalability of a solid-state host system and can operate at room temperature. So far, the optical cycle of the spin-polarization of the negatively charged nitrogen-vacancy (NV) center in diamond has represented the most studied prototypical example. The field is currently thriving with defects in several materials being studied in order to comply with more scalable fabrication processes, extend the coherence time, or enhance photon extraction. One key challenge remains the ability to generate stable defects while maintaining robust spin properties that dictate the quantum functionality.
Theoretical insights and broadly applicable models offer the opportunity to accelerate the experimental examination of candidate materials. We use predictive modeling techniques based on first-principles numerical simulations to provide a quantitative description of a broad class of materials to better guide and understand experimental activities. In particular, we use hierarchical modeling approaches, from density functional theory to many-body perturbation theory and multi-reference methods, harnessing pre-exascale computing, quantum computing, and artificial intelligence.
Photoluminescence spectra of point defects in semiconductors: Validation of first-principles calculations, Y. Jin, M. Govoni, G. Wolfowicz, S.E. Sullivan, F.J. Heremans, D.D. Awschalom, G. Galli, Phys. Rev. Mater. 5, 084603 (2021).Quantum Embedding Theory for Strongly-correlated States in Materials, H. Ma, N. Sheng, M. Govoni, G. Galli, J. Chem. Theory Comput. 17, 2116 (2021).
First-principles Studies of Strongly Correlated States in Defect Spin Qubits in Diamond, H. Ma, N. Sheng, M. Govoni, G. Galli, Phys. Chem. Chem. Phys. 22, 25522 (2020).
Quantum simulations of materials on near-term quantum computers, H. Ma, M. Govoni, G. Galli, npj Comput. Mater. 6, 85 (2020).
PyZFS: A Python package for first-principles calculations of zero-field splitting tensors, H. Ma, M. Govoni, G. Galli, J. Open Source Sofw. 5(47), 2160 (2020).
Fundamental principles for calculating charged defect ionization energies in ultrathin two-dimensional materials, T. J. Smart, F. Wu, M. Govoni, Y. Ping, Phys. Rev. Materials 2, 124002 (2018).
Designing defect-based qubit candidates in wide-gap binary semiconductors for solid-state quantum technologies, H. Seo, H. Ma, M. Govoni, G. Galli, Phys. Rev. Materials 1, 075002 (2017).
Design of defect spins in piezoelectric aluminum nitride for solid-state hybrid quantum technologies, H. Seo, M. Govoni, G. Galli, Sci. Reports 6, 20803 (2016).
Materials for Energy
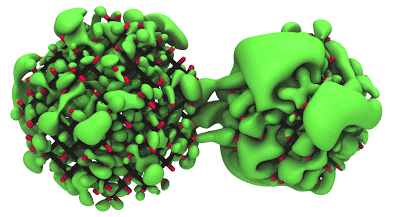
Study nanostructured materials for solar cell applications using electronic structure calculations based on density functional and many body perturbation theory
Nanostructured materials are emerging as tunable, earth-abundant and potentially non-toxic materials for solar energy conversion, light emission and optoelectronic applications. Theory and simulations are used to help interpret a growing body of complex measurements, and to predict optimal systems for harvesting solar photons, and materials with desired electronic transport properties.
The investigation of new materials that can extract, store and use energy in a more efficient way represents a key challenge of basic energy science research. Whether the aim is finding next generation photovoltaic materials, efficient light absorbers for water splitting or efficient rechargeable batteries, the answer is sought by studying the combined effect of photo-electro-chemical reactions that happen at the atomistic scale. We use a theoretical framework based on molecular dynamics and electronic structure simulations for large systems that can address different aspects of basic energy science research. Our aim is to gain an understanding of the basic reactions that govern fundamental excitations and the chemistry of the nanomaterials, simulating the microscopic mechanisms that rule the efficiency of energy operating devices.
The role of defects and excess surface charges at finite temperature for optimizing oxide photoabsorbers, M. Gerosa, F. Gygi, M. Govoni, G. Galli, Nature Materials 17, 1122 (2018).Carrier Multiplication in Silicon Nanocrystals: Theoretical Methodologies and Role of the Passivation, I. Marri, M. Govoni, S. Ossicini, Phys. Status Solidi C 14, 1700198 (2017).
First-principles calculations of electronic coupling effects in silicon nanocrystals: Influence on near band-edge states and on carrier multiplication processes, I. Marri, M. Govoni, S. Ossicini, Sol. Energ. Mat. Sol. C. 145,162 (2016).
Carrier multiplication in silicon nanocrystals: ab-initio results, I. Marri, M. Govoni, S. Ossicini, Beilstein J. Nanotechnol. 6, 343 (2015).
Red-Shifted Carrier Multiplication Energy Threshold and Exciton Recycling Mechanisms in Strongly Interacting Silicon Nanocrystals, I. Marri, M. Govoni, S. Ossicini, J. Am. Chem. Soc. 136, 13257 (2014).
Carrier multiplication between interacting nanocrystals for fostering silicon-based photovoltaics, M. Govoni, I. Marri, S. Ossicini, Nature Photonics 6, 672679 (2012).
Aqueous Solutions

Study solvation of simple ions, and water on surfaces in extended and confined media
First principles simulations of water are a challenging problem because one has to provide an accurate description of the different type of bonds present in water: intra-molecular covalent bonds, inter-molecular hydrogen bonds and van der Waals interactions. Additional difficult tasks include accounting for proton quantum effects, and efficiently sampling the complex potential energy surface of water.
In spite of these challenges, we are making progress in understanding solvation properties of simple ions, unraveling interactions between water and surfaces, and in predicting spectroscopic properties of aqueous systems and of water and ice under pressure. We mostly use ab initio molecular dynamics and many body perturbation theory.
Electron affinity of liquid water, A. Gaiduk, T.A. Pham, M. Govoni, F. Paesani, G. Galli, Nature Comm. 9, 247 (2018).Electronic structure of aqueous solutions: Bridging the gap between theory and experiments, T.A. Pham, M. Govoni, R. Seidel, S.E. Bradforth, E. Schwegler, G. Galli, Science Advances 3 (6), 1603210 (2017).
Photoelectron Spectra of Aqueous Solutions from First Principles, A.P. Gaiduk, M. Govoni, R. Seidel, J.H. Skone, B. Winter, G. Galli, J. Am. Chem. Soc. Commun. 138, 6912 (2016).
Methods and Codes Development

Develop methods and codes to understand and predict the properties of molecules and condensed systems
Method developments include finite-temperature electronic spectroscopies, development of dielectric-dependent hybrid functionals, and constrained DFT.
We lead the development of the WEST code, and contribute to the development of the Qbox code; in particular we focus on coupling first principles molecular dynamics with many body perturbation theory (GW, BSE, electron-phonon) methods, and with advanced sampling techniques (e.g., SSAGES). The combined used of ab initio molecular dynamics and electronic structure simulations on large systems opens the way to simulations that can be both descriptive and predictive, that is they can reproduce the experimental observations and also guide the research towards novel directions.
Together with the development of Python packages, particular effort is dedicated to the development of open source-software for quantum simulations that can be deployed on pre-exascale leadership class facilities, e.g., Argonne Leadership Computing Facility (ALCF), NERSC, and CINECA. We are currently expanding the simulations to include elements of artificial intelligence and quantum computing.
We also develop the open source software Qresp which facilitates the organization, annotation, and exploration of data presented in scientific papers.
Quantum Embedding Theory for Strongly-correlated States in Materials, H. Ma, N. Sheng, M. Govoni, G. Galli, J. Chem. Theory Comput. 17, 2116 (2021).Machine Learning Dielectric Screening for the Simulation of Excited State Propertiesof Molecules and Materials, S. Dong., M. Govoni, G. Galli, Chem. Sci. 12, 4970 (2021).
Code interoperability extends the scope of quantum simulations, M. Govoni, J. Whitmer, J. de Pablo, F. Gygi, G. Galli, npj Comput. Mater. 7, 32 (2021).
Quantum simulations of materials on near-term quantum computers, H. Ma, M. Govoni, G. Galli, npj Comput. Mater. 6, 85 (2020).
PyCDFT: A Python package for constrained density functional theory, H. Ma, W. Wang, S. Kim, M.-H. Cheng, M. Govoni, G. Galli, J. Comp. Chem. 41, 1859 (2020).
PyZFS: A Python package for first-principles calculations of zero-field splitting tensors, H. Ma, M. Govoni, G. Galli, J. Op. Source Softw. 5(45), 1945 (2020).
MatD3: A Database and Online Presentation Package for Research Data Supporting Materials Discovery, Design, and Dissemination, R. Laasner, X. Du, A. Tanikanti, C. Clayton, M. Govoni, G. Galli, M. Ropo, V. Blum, J. Op. Source Softw. 5(45), 1945 (2020).
Improving the efficiency of G0W0 calculations with approximate spectral decompositions of dielectric matrices, H. Yang, M. Govoni, G. Galli, J. Chem. Phys. 151, 224102 (2019).
Dielectric dependent hybrid functionals for heterogeneous materials, H. Zheng, M. Govoni, G. Galli, Phys. Rev. Mater. 3, 073803 (2019).
Finite field approach to solving the Bethe Salpeter equation, N.L. Nguyen, H. Ma, M. Govoni, F. Gygi, G. Galli, Phys. Rev. Lett. 122, 237402 (2019).
Qresp, a tool for curating, discovering and exploring reproducible scientific papers, M. Govoni, M. Munakami, A. Tanikanti, J. H. Skone, H. B. Runesha, F. Giberti, J. de Pablo, G. Galli, Sci. Data 6, 190002 (2019).
A Finite-field Approach for GW Calculations Beyond the Random Phase Approximation, H. Ma, M. Govoni, F. Gygi, G. Galli, J. Chem. Theory. Comput. 15, 154 (2019).
Coupling First-Principles Calculations of Electron-Electron and Electron-Phonon Scat-tering, and Applications to Carbon-Based Nanostructures, R.L. McAvoy, M. Govoni, G. Galli, J. Chem. Theory. Comput. 14, 6269 (2018).
Dielectric properties of condensed systems composed of fragments, D. Pan, M. Govoni, G. Galli, J. Chem. Phys. 149, 051101 (2018).
GW100: Comparison of methods and accuracy of results obtained with the WEST code, M. Govoni, G. Galli, J. Chem. Theory Comput. 14, 1895 (2018).
Performance and self-consistency of the generalized dielectric dependent hybrid functional, N. Brawand, M. Govoni, M. Vörös, G. Galli, J. Chem. Phys. 149, 051101 (2018).
Generalization of dielectric dependent hybrid functionals to finite systems, N. Brawand, M. Vörös, M. Govoni, G. Galli, Phys. Rev. X 6, 041002 (2017).
Implementation and Validation of Fully-Relativistic GW Calculations: Spin-Orbit Coupling in Molecules, Nanocrystals and Solids, P. Scherpelz, M. Govoni, I. Hamada, G. Galli, J. Chem. Theory Comput. 12, 3523 (2016).
Nonempirical range-separated hybrid functionals for solids and molecules, J.H. Skone, M. Govoni, G. Galli, Phys. Rev. B 93, 235106 (2016).
Large Scale GW Calculations, M. Govoni, G. Galli, J. Chem. Theory Comput. 11, 2680 (2015). Self-consistent hybrid functional for condensed systems, J.H. Skone, M. Govoni, G. Galli, Phys. Rev. B 89, 195112 (2014).
Auger recombination in Si and GaAs semiconductors: Ab initio results, M. Govoni, I. Marri, S. Ossicini, Phys. Rev. B 84, 075215 (2011).